|
|
|
"Formula Not Needed"
A common misconception is that the first step in identifying an unknown analyte from mass spectral data, is to determine its molecular formula. This video demonstrates the identification of thiazopyr from its MS/MS fragmentation data. Using our Excel Add-in, this compound was identified from its accurate mass and its subformulas. "Formula not needed"
"How well does the Rational Numbers Excel Add-In work?"
Although our Add-In is designed to be used interactively, we recently analyzed thousands of mass spectra in an automated way. We chose to analyze spectra in MassBank provided by EAWAG and LCSB which are all spectra of outstanding quality. When used interactively, our Excel Add-In will often detect and flag odd-electron fragment ions even if the user had not selected "Odd e SubFrags OK"; it will also often flag fluorine-containing compounds even if the fluorine count maximum is set at zero; and it will advise on whether there are too many possible formulas for the current window settings. To run the thousands of spectra automatically, it was necessary to always allow odd-electron subfragment formulas. These thousands of analyses are displayed sorted by Pubchem Compound ID (PCID). The results are also searchable by PCID. See how the Excel Add-In worked with some compounds that you are interested in. (References to the original MassBank data are listed toward the bottom of the DataInput worksheets.)
While you ideally would like to obtain only modular structures that exactly correspond to the analyte that produced the spectral data, you can realistically only expect that the Excel Add-In produces modular structures that are consistent with the accurate-mass fragmentation data.
"Out-of-Context"
Politicians are often complaining that remarks that they previously made have been taken "out-of-context"; reporters who quoted just one or two sentences were perhaps distorting their intended message. Like quotes, mass spectra always have to be interpreted in context. An individual fragment ion mass tells us very little.
NMR spectra have characteristic functional group shifts which are easily distingishable and fairly predictable for small molecules. Likewise, in the IR spectra of small molecules, many functional groups have consistent and characteristic signals. For example, the C=O stretch of a carbonyl group almost always appears at around 1700cm-1. Hence, known correlations with chemical structures of characteristic chemical shifts in an NMR spectrum or of the band wavenumber positions in IR can be used to help identify an analyte.
Unlike the signals in NMR or IR spectra, a fragment mass observed in a mass spectrum has no frame of reference. Each fragment mass must be interpreted in the context of all other masses. Very few individual masses and mass differences (e.g. loss of methanol, 32.0262 daltons) can be directly related to a particular substructure of an analyte.
As an example, suppose a mass spectrum had a fragment ion with a mass of 67.0542 daltons in positive ion mode. The most likely formula is C5H7+. The neutralized fragment ion mass, used in partitioning with the Excel Add-In, could then generate some modular structures where one subfragment has the formula C5H6. This is a combination of eleven atoms, but combinations have no ordering. Furthermore, the number of hydrogens in a subformula often differs from the number of hydrogens in the corresponding substructure. A variety of molecules have substructures that can give exactly the same fragment ion mass and the same subformula. Here are six analytes that have at least one C5H6 subformula. Five of the six molecules in the figure have substructures that differ from the corresponding subformulas in the number of hydrogen atoms. The figure also shows the other subformulas in the same partition which provide some "context".
"YouTube Video: Cimetidine MS/MS with the Excel Add-In"
Cimetidine is a drug used to treat both ulcers and acid reflux disease. I recently created a YouTube video that describes a procedure for inputting data from MassBank into our Excel Add-In for testing out the Excel Add-In or for checking spectra in MassBank. This video used cimetidine data. You can find that YouTube video here. Cimetidine MS/MS data from MassBank in positive ion mode that was analyzed in more detail using the Excel Add-In is here.
See many other examples of our Excel Add-In.
"Seeing what is not there"
Fluorine and phosphorus atoms have no natural isotopes. So when calculating the isotope pattern of a molecule based on natural isotope abundances, the number of fluorine and phosphorus atoms in a molecule can be completely ignored. These two elements have no effect whatsoever on the estimated isotope patterns.
If we plot the average percent relative abundance of the first isotope (M+1) of the 250000 common molecules in our SPS database (low resolution) against the integral mass of the molecules, the average percentage closely fits a straight line. The plot is shown here and it has this linear equation: average relative % = 0.0566 * mass + 0.6037. If we then create a rough "rule of thumb", the average relative % of the first isotope would be the mass of the molecule divided by 100 and then multiplied by 6%. Therefore, a compound of mass 500 daltons would be expected to roughly have a relative % abundance of 500/100 * 6% = 30%.
Perfluorooctanesulfonic acid has an exact mass of 499.9375 daltons and so, based on this mass, it might be expected that its first isotope would have a relative abundance of roughly 30% - assuming it is an "average molecule". However, based on its molecular formula (C8HF17O3S), the estimated relative abundance of the first isotope is only 9%.
At low resolution, the first isotope is dominated by carbon-13. While fluorine has no effect whatsoever on isotope ratios, it does constitute a significant percentage of the mass in this perfluoro compound (65%) and fluorine therefore exerts its influence on the isotope ratio indirectly by reducing the percentage of carbon to 19% in this molecule. Perfluoro compounds have first isotopes with relative intensities that are less than half of the expected average relative intensity.
Chlorine and bromine can also reduce both the percent carbon and the relative intensity of the first isotope in the same way, but these two elements have an intense M+2 isotope. On the other hand, it is more difficult to reduce the mass percentage of carbon in a molecule with other elements like oxygen. For example, raffinose (C18H32O16, 504.169 daltons) is 43% carbon and it has an estimated relative abundance of the first isotope of 20%, which is more than half of the expected average relative intensity.
What is the difference in nomenclature between an "estimated isotope pattern" and a "theoretical isotope pattern"? The difference is described in Dan's book, "Mass Spectrometry by the Numbers".
"HF + CO2 → CH4OS "
Molecules with a methyl sulfinyl group will often lose a CH4OS moiety (mass 63.9983 daltons) under MS/MS conditions. An example was described in "Mass Spectrometry by the Numbers" pp 101-103, a metabolite of clindamycin where the sulfur atom of a methyl thioether had been oxidized to a methyl sulfinyl group.
Another compound that loses virtually the same mass is flunixine (see examples); it loses 63.9960 daltons in negative ion mode. However, flunixine does not even have a sulfur atom in its structure. This loss of 64 in flunixine is actually two separate losses: HF and CO2 (63.996 daltons). The Excel Add-In looks for 134 different combinations of two small subfragment masses that sum to another small subfragment mass. Every simple mass difference of 70 daltons or less is checked to see if it can be explained better as a sum of two smaller masses. That is why no partition of flunixine in negative ion mode uses the mass difference of 64 as a subfragment mass and assigns it as CH4SO.
The Excel Add-In also checks 21 masses to determine if the MS/MS spectrum is contaminated with clusters of an inorganic cation. It also checks for some common organic interferences like phthalates, stearic acid, and palmitic acid. Lastly, it checks for "extraterrestrial ions" that have mass defects that are really not possible (e.g 170.22 mDa).
These checks can all be done without the Excel Add-In by carefully reviewing your data. But life is short; purchase your copy of the Excel Add-In today.
"Natural Variations in Isotopic Abundances"
Some mass spectrometrists still believe in the concept of "theoretical isotope ratios" despite factors that can significantly affect isotope ratios such as sample size and natural variations of isotopic abundances. These factors are discussed in Chapter 2 of my book,"Mass Spectrometry by the Numbers". The Commission on Isotopic Abundances and Atomic Weights has a noteworthy graph on oxygen isotopic distribution (as atomic weight) in US river waters. Regional variations in isotope ratios have also recently been used to discover that many of the human bones buried about 5000 years ago at Stonehenge were bones from people who did not live in the surrounding area, but more likely bones of people who lived in west Wales. This work was based on the regional variation of strontium isotopes in Great Britain.
Opposite World
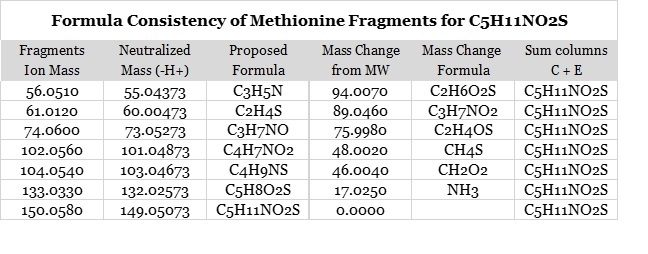
When the formula consistency checkbox of the Excel Add-In is selected (default), all possible formulas are checked for consistency. Using the example of the methionine positive ion ms2 spectrum found in MassBank (PB000441), one proposed formula for the whole molecule is C5H11NO2S. If that proposed formula is correct, it follows that all of the fragment ions of that molecule must have compositions that are subsets of C5H11NO2S. This is always true unless that spectrum has extraneous ions from co-eluting analytes; fortunately extraneous ions are not frequently a problem.
In addition, each neutralized fragment ion and its mass difference from the molecular weight can be viewed as a two-cell partition of the molecular weight. This means that the mass difference between the molecular weight and each neutralized fragment ion must also correspond to a formula that is a subset of the proposed molecular formula C5H11NO2S. This is shown in the Formula Consistency Table.
In an opposite world thought experiment, the mass of a proton can be added to each mass in the fourth column. This creates a new set of masses, together with the protonated molecular ion, that can be considered the "difference spectrum". The intensity of each ion in the difference spectrum would be the intensity of its complementary ion. We can then analyze both the original spectrum and its difference spectrum with the Rational Numbers Excel Add-In. The results for the first partition are shown here for the original spectrum and here for the difference spectrum. In opposite world, both the scores and the subfragment compositions are identical, but the correct alignment is not found because mass differences can often combine subfragments that are not connected, whereas fragment ions are almost always subfragments or connected groups of subfragments. (The required isotope data was taken from Massbank Spectrum KNA00300.)
Base Rates
An earlier blog posting, "Divide and Conquer", pointed out that as the mass of a molecule or subfragment decreases - the number of possible formulas decreases. This idea was originally described by Quincy Wu. Based on this concept, by dividing an unknown molecule into smaller subfragments by using our Excel Add-In, we can considerably limit the formulas possible for the whole molecule.
There is a second factor that should also be taken into consideration. Consider a molecule like chloramphenicol (Pubchem CID 5959). Chloramphenicol has the formula C11H12Cl2N2O5, which is not very unusual. The structure of chloramphenicol has a carbon atom with 2 chlorine atoms at one end and the nitro group at the other end. If we consider possible formulas of subfragments of an unknown molecule with an overall formula of C11H12Cl2N2O5, how plausible is the subformula CHNO2Cl2 for a subfragment of mass 128.9384? CHNO2Cl2 does not violate any chemical rules (e.g. valence).
To study this, I divided up 230000 common compounds having masses between 100 and 680 daltons into 4 subgroups of atoms and collected the heavy atom (non-hydrogen atom) compositions and the frequency at which each composition occurred. CNO2Cl2 never occurred. Logically, as the subfragment mass and number of carbons increase, it becomes much more likely to see a heteroatom combination such as NO2Cl2 in the same part of a molecule. For example, the heavy atom composition C11NO2Cl2 in a single subgroup occurred in 539 compounds. (An example of a molecule with this subgroup is Pubchem CID 2747401.)
Overall, there were 10472 different heavy atom compositions found for the subgroups with a mass of 250 Daltons or less. The three most common heavy atom compositions were C2 (found in 202407 molecules), CO (found in 187438 molecules), and C3 (found in 183742 molecules). Some heavy atom compositions are very rare; 14.9% of the compositions (1560) were each found in only one molecule.
The BaseRate() function in the Excel Add-In will return the base rate (on a scale of 1 to 99) that describes the probability that any given heavy atom subfragment composition will occur in an analyte - if you knew nothing else at all about the analyte. Formulas with a base rate of zero are not considered as possible elemental compositions of subfragments. The BaseRate() function is limited to masses of 250 daltons and less. Like the other functions in the Add-In, the BaseRate() function can fill down and fill across.
The "Relative Feasibility" score in partitions produced with the Excel Add-In are calculated by dividing the base rate of the subfragment formula with the lowest baserate in each row by the average subfragment mass error of that row. There is also a weighing factor for the number of subfragments in the partition. Thus this score takes base rates into account.
Incorporating the BaseRate() function, the "Formula Restriction" parameter on the DataInput worksheet can be used to rule out the less common subfragment formulas.
Ambiguity, Chance, and Mass Spectral Identification
Identifying unknown compounds from mass spectral data is tackling a problem with no clear solution. The Excel Add-In should be viewed as a tool that speeds up the processing of the spectral data ? an optimized calculator for mass spectrometry. The results will lead to multiple possibilities that are consistent with the data, although not all of the possibiilties will be consistent with the structure of the compound that produced the data. Because of the inherent ambiguity of mass spectral data, the objective should be to generate a variety of hypothetical structures consistent with the data.
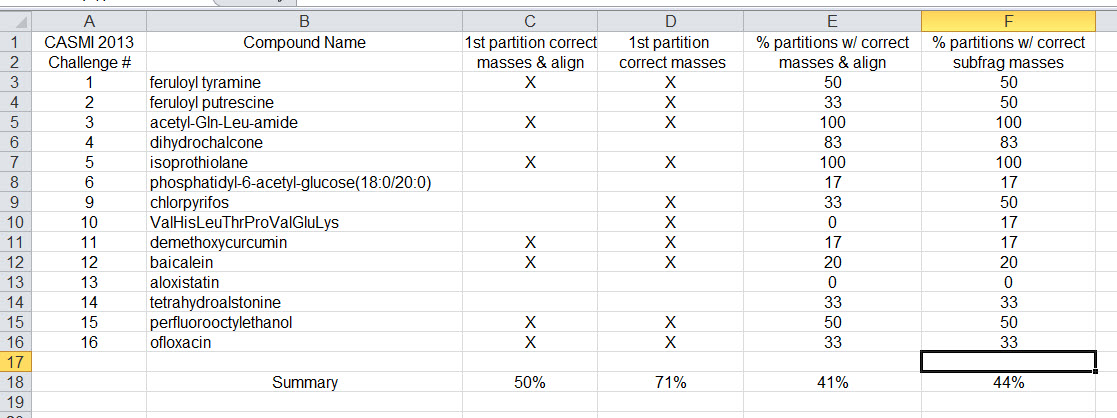
I recently analyzed the 16 compounds from the CASMI 2013 Challenge with the latest version of the Rational Numbers Excel Add-In. These are compounds and datasets that were not cherry-picked consciously or unconsciously. Of the 16 compounds, the Challenge 7 dataset (error > 11 millidaltons) is excluded here because it is not really accurate mass - here defined as data where 95% of the fragment ions are within 5 millidaltons of their theoretical mass. There were 15 compounds left.
There are multiple obstacles to identifying compounds from accurate-mass fragmentation data. First, some compounds do not fragment well and perhaps give only one fragment ion. In the CASMI 2013 Challenge, perhaps because it was a contest of sorts, all of the given data sets had multiple fragment ions.
Challenge 8 had a molecular weight > 999, which is the upper limit of the Excel Add-In. So 7 % of the compounds in this set were too massive to analyze with the Excel Add-In.
In analyzing the spectrum of an unknown, one must assume that the spectrum is a spectrum of a molecule that has had no major rearrangements; otherwise it is not possible to draw any conclusions. Fortunately, most fragment ions arising from rearrangements are very minor, and so the Excel Add-In ignores fragment ions with intensities less than 1.5%. Of the remaining fragment ions, only the protonated or deprotonated molecule and the 11 most intense fragment ions are saved. Three of the compounds (20%) had fairly significant rearrangements: feruloyl putrescine (Challenge 2), baicalein (Challenge 12), and aloxistatin (Challenge 13).
There are basically two types of rearrangements. Some compounds will lose a group from a middle position (e.g loss of SO2 from the center of a sulfanilamide) or will have a chemical group migrate from one position in a molecule to another. This will often lead to an incorrect alignment or no alignment, but the subfragment masses and compositions will be correct. The second type of rearrangement changes the overall structure of the molecule. When the base peak in the spectral data originates from this type of rearrangement, this rearrangement makes it almost impossible to identify the compound that produced the data.
Feruloyl putrescine had a subfragment that was extruded from the middle (ketene in this case) giving a fragment ion at 206.1163 with a 16% relative intensity. Although a subfragment was lost from the middle, its mass corresponded to the molecular structure. The subfragment masses were therefore correct, but the alignment of the subfragments in space were self-contradictory or incorrect in the two partitions where the 206 ion was assigned. This was the case with feruloyl putrescine Partitions 1 and 2.
In contrast, the base peak in the MS/MS spectrum of aloxistatin was the result of a major rearrangement that considerably altered the molecular structure. All six partitions of aloxistatin that were found by the Excel Add-In were incorrect. Although the spectrum of the flavone baicalein had multiple fragment ions resulting from rearrangements, in this case the three largest fragment ions all corresponded to the structure. As a result, the first and highest scoring partition correlated to the structure of baicalein but the remainder of the partitions did not. (Rearrangements of flavones: Fabre, Rustan, Hoffman, and Quetin-Leclercq in JASMS 2001, 12, pp 707-715).
A more common problem in mass spectral identification is two pieces of a molecule having the same composition but being perceived as only one piece. With the Excel Add-In, this generally leads to contradictory assignments which in turn leads to either no alignment or an incorrect alignment of the subfragments. A good example is Partition 1 of leucine enkephalin (not in this Challenge 2013 dataset) which has two glycine amino acid moieties but only one subfragment of mass 57 that is being used for both; this led to no alignment being assigned. None of the 15 Challenge 2013 compounds had this problem, althougn it is more common than rearrangements. Duplicated compositions within a molecule also explains why compounds that have very few heteroatoms and multiple common pieces (e.g. steroids, prostaglandins) are usually more difficult to identify than compounds with many different heteroatoms, even though having more heteroatoms increases the number of possible molecular formulas.
Less common is two subfragments explaining a fragment ion by chance. In ValHisLeuThrProValGluLys (Challenge 10, Partition 1), although the subfragment masses are all correct, the 432 fragment mass (450 minus water) was assigned as the sum of the 196 +236 subfragments. The elemental composition is the same. However, this is incorrect and led to no alignment being assigned. The subfragment masses and compositions were all correct.
The last identification problem is the ?Snow White and the Seven Dwarfs? problem. In these datasets, solutions are found that are consistent with both the data and the structure, but one of the subfragments (?Snow White?) is much larger than the others (?Dwarfs?). For example, baicalein (Challenge 12) and perfluorooctylethanol (Challenge 15) yielded partitions where one of the subfragment masses accounted for over 80% of the mass of the whole compound. Perfluorooctylethanol (Partition 1) had 4 HF subfragments and one 384 subfragment with a very large number of possible compositions. The Excel Add-In uses a mass variance factor that favors subfragments of average size, but that does not always help. One solution in these situations is to follow up with MS3 on the fragment ion corresponding to the Snow White subfragment.
Overall, for the data sets of the 15 compounds from CASMI Challenge 2013, 41% of all partitions corresponded to the molecule in both alignment and subfragment composition and 44% of all partitions had a correct subfragment composition. For the first partition of each dataset (top scoring partition) 50% of the partitions corresponded to the molecule in both alignment and subfragment composition and 71% of the partitions had correct subfragment compositions. View the summary here.
In many cases multiple elemental compositions were possible; the number of possible compositions varies widely based on the accuracy of the data and whether fluorine can be eliminated from consideration (e.g. natural products). All of the individual results and many other examples are shown here.
Think Outside the Cone
Before analyzing the data, the Excel® Add-In always does four checks on the data, looking for two types of fragment ions: the "extraterrestrial" and the "obtrusive" . The Add-In checks the masses of the 12 most intense fragment ions before it proceeds.
Extraterrestrial ions have masses that do not make any sense for the elements C, H, N, O, S, P, F, Cl, and Br. For example, hydrocarbons have the greatest relative mass defect, because hydrogen has by far the greatest mass defect of all the elements in our universe and hydrocarbons have the highest percentage of hydrogen atoms. Pubchem CID 8182, dodecane, has a nominal mass of 170 and a mass defect of 203.4 millidaltons. Therefore, no compound of nominal mass 170 or less can have a mass defect greater than 203.4 millidaltons + the maximum error window of 5 millidaltons. If the dataset has a fragment ion at 170 with a mass defect of 210 mDa there is a problem with that data point. Plotting hydrocarbon nominal mass against mass defect; a line is obtained; each CH2 unit adds 1.118 millidaltons per nominal dalton of mass.
A similar, but more complicated, argument can be made for calculating the smallest (or most negative) mass defect for a given nominal mass. A CCl2 unit will decrease the defect by 0.760 per nominal dalton of mass. Plotting this, again a line is obtained. (There are a few exceptions with bromine that the Excel Add-In takes into consideration.)
If one plots the maximum positive ion defect versus the nominal mass and the minimum negative ion defect versus the nominal mass on the same graph, the cone of extraterrestrial mass defects is obtained. This cone has a vertex at about 525 daltons; for masses greater than 525 any mass defect is possible.
No fragment ion should be found with a mass defect inside the cone. But this concept also applies to losses: the mass defect of the difference between the mass of the molecular ion and the mass of any fragment ion also must be outside the cone. This is a second check on the dataset.
There are also two mass loss ranges that are excluded, based on the masses of the elements C, H, N, O, S, P, F, Cl, and Br. The masses between the mass of 4 and 14 can be excluded as well as the masses between the mass of HF (20.0062) and cyanide (26.0031), taking into account the maximum error of 5 mDa. This is a third check on the dataset.
Obtrusive ions usually creep into a spectrum from the mobile phase. By chance, some combination of mobile phase components and impurities in the mobile phase form an adduct that sums up to a mass close to the mass of the desired precursor ion. The Excel Add-In looks for formate, acetate, stearate, palmitate, and trifluoroacetate in negative ion mode. In the positive ion mode, the Excel Add-In looks for the 149 ion of phthalates. Obtrusive ions are flagged, but may very well belong in the spectrum.
Artifacts can also arise if nitrogen is used as the collision gas. Although nitrogen is usually regarded as an inert gas, it will react with arylium ions. In April 2015, researchers at NIST, Yuxue Liang, Pedatsur Neta, Yamil Sim?n-Manso,and Stephen E. Stein, published a paper in Rapid Communications in Mass Spectrometry, Volume 29(7)pp 629-636, where they demonstrated conclusively that some ions in MS/MS spectra that could not have arisen from the precursor ion, can be explained by reaction of some initially formed arylium ions with nitrogen to form diazonium cations. An example of the problem is the MassBank spectrum of 2-toluenesulfonamide (www.massbank.jp spectrum # EQ360501). There is a very intense fragment ion (86%) at 119 daltons which is due to the reaction of the benzyl cation at 91 with nitrogen to form a diazonium ion with a mass difference of N2. This would make the correct identification of such a compound from this MS/MS spectrum very difficult; real N2 mass differences are observed in the spectrum of compounds such as benzotriazoles (e.g. massbank spectrum # EA016613). This rather serious problem can be avoided completely by using argon as the collision gas. Although argon is relatively expensive, very little collision gas is actually used if there are no leaks.
Seeing Red
The color specification for many APIs is "white" or "off-white" leading to off-specification material when colorants are present. The eye is very sensitive to color and these colored impurities usually are present at trace levels and often not closely related to the drug molecule in structure. Because of its sensitivity, mass spectrometry is often called upon to identify colorants in APIs.
This is very challenging. Even though LCMS is extremely sensitive, it is usually necessary to concentrate the colorant prior to LCMS. For small molecule drugs under 500 daltons and soluble in methanol, one approach that may work is ultrafiltration through regenerated cellulose membranes. A concentrated solution of the drug in methanol is filtered through a 1 kDa-cutoff regenerated cellulose membrane using a stirred cell ultrafiltration unit. Ideally, a large volume stirred cell (400 mL) is used. The initial concentrate can be washed with additional pure methanol to remove more of the API.
The underlying principle is that colorants are highly conjugated molecules. This conjugation leads to rigid and very planar molecules which have a relatively large cross-section, thus hindering their passage through the 1 kDa-cutoff UF membrane relative to colorless drugs.
"Inside-Outside"
The subfragment compositions of the assigned fragments is a major contributor to the spatial alignment. For example, if fragment A-B is assigned and fragment B-C is assigned, it follows that B is between A and C. But other factors besides the fragment assignments can also be helpful.
Subfragments that have only one point of attachment must be on the outside. These one-handed subfragments include HF, HCl, HBr, small saturated alcohols (e.g. ethanol), and alkenes with only one double bond (e.g. isobutene). The Excel Add-In forces these one-handed subfragments to outside positions.
A logical assumption is that "outside" pieces of the molecule will often show up directly as neutral losses from the protonated or deprotonated molecular ion. So a molecule aligned as A-B-C-D might be expected to lose subfragment A or D. Conversely, loss of B or C would not be expected; organic chemists would call this an extrusion reaction. An extrusion is a type of rearrangement that does not affect subfragment masses or compositions, but will often lead to an incorrect alignment. Other rearrangements (e.g. migration of a methyl or benzyl group to another location on the molecule) usually lead to incorrect compositions of subfragments (e.g. bosentan).
The Excel Add-In always makes the "Inside-Outside" assumption that internal subfragments will not be lost directly from the whole molecule. Thus any exceptions to this rule will be excluded as possible alignments. Fortunately, extrusions are rare.
Sulfamethoxazole MassBank (http://www.massbank.jp) spectrum EA029801 is an example of a type of compound that is probably the most common exception to the "Inside-Outside" assumption. Sulfonamides will almost always lose a mass of 63.9619 (SO2) and/or 65.9776 (H2SO2 or SO2 and H2) from the protonated molecular ion by extrusion. Spectrum EA029801 shows both the loss of 64 and 66. In analyzing this spectrum of sulfamethoxazole, the Excel Add-In selects the SO2 as a subfragment rather than the H2SO2 because using that subfragment mass gives higher overall partition scores. In all five of the partitions found that included the SO2 subfragment, that subfragment was always placed incorrectly in an outside position. If the Excel Add-In macro forced the SO2 subfragment to be in a middle position, it is possible that no possible alignment would be found and that partition might then be dropped from consideration.
Some other small subfragments that may be extruded are ketene (C2H2O) (although most commonly found on the outside as a loss from acetate or acetamides) and CO2 from a carbobenzyloxy blocked amine.
View a comparison of dozens of mass spectral data sets analyzed with both the Rational Numbers™ Excel Add-In and the Rational Numbers™ Search software here.
Size Does Matter
Large subfragments often have too many possible elemental formulas - while very small subfragments (e.g water, ammonia) are not very informative in identifying unknown compounds.
The Excel Ad-In slightly favors medium size subfragments. The average mass of the subfragments is calculated from the molecular weight. Then, based on the magnitude of the mass difference between each individual subfragment and the average mass, a slight penalty is assessed. This penalty is not large enough to preclude very small subfragments or very large subfragments from being used, but it generally helps to produce sets of subfragments that are more useful for identification work.
Two spectra of compounds of approximately 500 dalton MW can be used to illustrate this.
The first example is the analysis of MassBank (http://www.massbank.jp) spectrum MSJ00027 of 2-(perfluorooctyl)ethanol. Despite the penalty that is intended to reduce the number of small subfragments, the highest scoring partition of 5 subfragments (Partition 1) had 4 very small subfragments, each composed of HF. Since the molecular weight is 464 daltons, the remaining large subfragment had a mass of 384 daltons. Despite using a fairly tight error window of 3 millidaltons, the 384 dalton subfragment had 107 possible elemental compositions.
The second example is glycovir (MW 499) and here a 4 millidalton error window was used for the analysis of the mass spectral data with the Excel Add-In. The spectrum of glycovir has four mass differences of 18 (H2O), but neither of the two partitions with the highest score (102, Partitions 1 and 2) had a subfragment of water. In fact, the first two partitions found had a range of subfragment masses between 70 and 176. The second partition was very representative of the structure; it had only two possible molecular formulas - including the correct formula - and only one possible alignment.
Odd Couples
Odd-electron subfragments always come in pairs. Furthermore, these odd-electron subfragments are usually attached together in the molecule; the only exception is compounds with major rearrangements - such as bosentan, with these Excel Add-In partitions. To find odd-electron pairs of subfragments with the Excel Add-In, use the RDE() function. All odd-electron subfragments have ring and double bond equivalents with a half (e.g 2.5); even electron subfragments have integral RDEs (e.g 2.0, 3.0 etc.).
A good example of a partition having a pair of odd-electron subfragments is Partition 1 from the analysis of MassBank spectrum KOX00157 of chloramphenicol with the Excel Add-In. The nitrogen dioxide subfragment (NO2, 45.9929) is odd-electron and it is attached directly to the aryl group - the other odd-electron subfragment. (The Excel Add-In does not force odd electron subfragments to be adjacent.)
Divide and Conquer
The Excel Add-In analysis of glycovir, with a mass error window setting of 4 millidaltons, had only two possible formulas for Partition 2: C23H46NO9F and C26H45NO8. By analyzing the molecular ion's mass (theoretical 500.3218 daltons) with a typical mass spectral program to find all possible formulas within 4 millidaltons of 500.3218 while excluding chlorine, bromine, and silicon, it appears that there should be 106 possible formulas.
The reason why the Excel Add-In found 104 fewer possible formulas is that both the masses and the elemental compositions of subfragments that compose partitions are mutually exclusive. In Partition 2 there are 3 subfragments of mass 88.0526; with a 4 millidalton error window only one formula is possible at 88.0526 (C4H8O2). The one subfragment of 70.0420 also has only one possible formula (C4H6O). These four masses can be excluded from the 499.3174 total mass calculated by the Excel Add-In from the data. By subtracting these four subfragments out from the total (499.3174 - 3 * 88.0526 - 70.0420 = 165.1176) the remaining (fifth subfragment) has only 2 possibilities: C7H16NO2F and C10H15NO.
The classic paper on this phenomena is Quincey Wu's "Basket-in-a-Basket" paper (Wu,Q. Anal. Chem. 1998, 70, 865-872). Instead of using the mathematical approach employed by the Excel Add-In to find partitions and thus limit the possible molecular formulas, Quincey used accurate mass MS5.
Some Masses Are More Equal Than Others
Three types of masses are used by the Excel Add-In as potential masses of subfragments. First, there are the masses of neutralized fragment ions. Then there are mass differences between pairs of fragment ions. And finally there are sum differences which are the sums of two neutralized fragment ions minus a third neutralized fragment ion. A twelve ion spectrum would have 298 masses and mass differences.
Below is the spectrum listing of the positive ion MS/MS spectrum KOX00526 of puromycin which has 8 ions.
118.0657 22.0000
121.0653 23.0000
150.0919 291.0000
160.0610 54.0000
164.0936 165.0000
292.1185 31.0000
309.1450 459.0000
472.2308 999.0000
We can compute all possible mass differences. Many of the mass differences do not correspond to possible subfragment masses, and it is difficult to distinguish the meaningful from the meaningless. For example the 309.1450 fragment ion above of puromycin actually has the composition C15H20N2O5. The 164.0936 fragment ion has the composition C7H9N5. From the numbers, the difference of 145.0514 might appear to have an elemental composition of C9H7NO (145.0528), but based on the known compositions of the ions, the 145.0514 mass difference actually corresponds to C8H11N-3 - which is meaningless.
Since no rearrangements are assumed, the most reliable subfragment masses are the neutralized fragment ions because these groups of atoms must be connected together in the molecule. These are called primary masses. Puromycin Partition 1 is very unusual in that all three of its subfragments have masses that are primary masses.
The next most reliable set of masses are small direct losses from the molecular ion (e.g a loss of 17 or 42). These are also considered primary masses. It is necessary to exclude losses that may actually be sums of two or more direct losses from different parts of the unknown molecule. For example, if a molecule would have losses of 17, 42, and 59 from the molecular ion, then the 59 mass would not be considered a primary mass.
Partitions that use primary masses get a slight bonus in scoring for every primary mass used as a subfragment mass. There are not many primary masses.
Secondary masses are differences between two fragment ions. As mentioned previously, these may or may not correspond to connected groups of atoms in the structure of the unknown compound. Partitions that use secondary masses get no adjustment in scoring for every secondary mass used as a subfragment mass. There are many secondary masses.
Some mass numbers may appear more than once. For example, the 149 subfragment mass of puromycin is 150.0919 - proton = 149.0846. It is also 309.1450 - 160.0610 = 149.0840. When multiple independent values are obtained, then the values are averaged together. If a mass is found more than once, then it gets the most favorable classification; the 149 mass here would be considered a primary mass.
Tertiary masses are sum/differences between neutralized fragment masses. The spectra of a small percentage of compounds have "hidden subfragments" that are not simple differences. For example, triclocarban (MassBank EA298809) has two major neutralized fragment ions at 127.0187 and 160.9797 and a neutralized molecular ion at 313.9777. Adding 127.0187 to 160.9797 gives 287.9985. Subtracting 287.9985 from 313.9777 gives 25.9793, the mass of the "hidden subfragment", which here corresponds to a COH-2 (calculated as 25.9793). The partition masses found in this way are: 127.0187 + 25.9793+ 160.9797 = 313.9777. (In this spectrum there are also very small fragment ions at 154.0053 and 187.9663, if these masses were greater than 1%, then the 26 subfragment mass might actually be considered a secondary mass.)
Tertiary masses are very often meaningless. Partitions that use tertiary masses get a penalty in scoring for every tertiary mass used as a subfragment mass.
"Degassers and MS/MS Spectra"
In contrast to in-source CID MS spectra and MSe type spectra, LC-MS/MS spectra are usually thought to be very pure spectra because of the combination of HPLC separation and the additional selectivity of the first mass spectral analyzer. However, this is not always the reality.
Vacuum degassers can be a source of spectral contamination. As an example, MassBank MS/MS spectrum (http://www.massbank.jp) KOX00065 of trans-aconitic acid PubChem 444212 is actually the spectrum of three compounds: trans-aconitic acid, trifluoroacetic acid (TFA), and acetic acid (HOAc). The exact mass of trans-aconitic acid (C6H6O6) is 174.0164; the exact mass of the complex of TFA and HOAc is 174.0140. Because the ion selection in the first analyzer is unit mass, a combination spectrum is obtained here accounting for the extraneous ions at 59 (CH3COO-), 69 (CF3-) and 113 (CF3COO-). Vacuum degassers are very convenient accessories for HPLC systems, but these units are often reservoirs of trifluoroacetic acid. Once contaminated, it is virtually impossible to remove TFA from vacuum degassers. In addition to interfering by chance with MS/MS spectra as illustrated here, the intense 113 ion in negative mode can suppress the ionization of compounds of interest. Whenever possible, bypass the degasser when using TFA in an LCMS system.
The KOX00065 data was analyzed with the Rational Numbers search program both with and without the TFA ions at 69 and 113. The Rational Numbers search program found aconitic acid as the best match in both cases but could not distinguish trans and cis isomers. Run time was 2 to 3 seconds for the search software. The data was also analyzed with the Excel Add-In both with and without the TFA ions at 69 and 113. Without the extraneous ions, the only two partitions found both had very high scores, attributed to the excellent mass accuracy of the data. With the two extraneous ions included, the scores of the two correct partitions (partitions 3 and 5) were much lower. Although these extraneous ions are fortunately not very common, the Excel Add-In checks for some of the problematic extraneous ions that may be present in spectra at the beginning of every analysis.
"High-Throughput Identification"
This "spectrum" is actually a set of 28 positive ion mode MS/MS spectra. The search software can now input a single mgf (Mascot) formatted chromatogram and analyze all of the MS/MS spectra in the chromatogram. This search approach is so fast that the identifications can be made as rapidly as you can generate MS/MS spectra on your instrument. Please watch the 10 minute YouTube video here.
"Ambiguity and Chance"
Why are many of the partitions found using the Excel Add-In inconsistent with the structure of the compound that generated the mass spectral data, even when there were no rearrangements? One example is the positive ion mode MS/MS spectrum of vecuronium bromide found in massbank.jp. Partitions 3 and 5 of the Excel Add-In, are both consistent with the structure, although both partitions have a molecular formula with one less hydrogen typical of quaternary compounds. If we compare partition 3 (consistent with the structure) to partition 2 (inconsistent), the partition scores are 72 for both. In addition, the overall scores (which include mass accuracy and comparison of the isotope ratios) are almost identical. Both partitions are derived by logical induction from the mass spectral data and are consistent with the data. Based on identical partition scores, the two solutions appear to be equally probable.
Not every solution that is consistent with the data will be consistent with the molecular structure. There is rarely enough information in a mass spectrum to derive a single correct structural formula. Even correct answers (e.g. partition 3 here) are really just blocks of elements (combinations). The arrangement of the atoms in each subfragment (permutations) are mental constructs except for very small compositions such as H2O or CH4O. Ambiguity and chance make mass spectrometry interesting. The Excel Add-In should not be viewed as a panacea, but rather as a tool to rapidly sort through mass spectral data and find ways of viewing the data that are consistent with the data. Analysis of the data with the Excel Add-In took 72 seconds.
The Rational Numbers search program found vecuronium bromide as the only match (results here) despite the apparent molecular weight discrepancy. Quaternary compounds are stored in the database one hydrogen mass down from their true exact mass so that they will be found easily. Run time was 2 to 3 seconds for the search software.
"Convolution Confusion"
The spectrum here, a positive ion mode MassBank (http://www.massbank.jp) KOX00689 MS/MS spectrum of cystathionine, has two interesting fragment ions. From the structure of cystathionine, it is very difficult to explain the loss of 46.0193 daltons from the precursor ion and also the 149.0234 fragment ion. This 46 mass difference appears to be too great for CH2O2 (formic acid, 46.0055) which would be a logical loss. If we examine another massbank spectrum of the same compound, ML003001, the loss of 46 is not there, and the large fragment ion at 149 is also missing. This might seem to be an effect of collision energy on the fragmentation, but in reality spectrum KOX00689 is the combined spectrum of two compounds: cystathionine and diethyl phthalate. Although there is no MS/MS spectrum of diethyl phthalate in massbank.jp, its EI spectrum JP005812 has major fragment ions at both 149 and 177 daltons. Both diethyl phthalate and cystathionine have a molecular weight of 222 daltons. Since diethyl phthalate is ubiquitous, it is very often found in mobile phases. Although the instrument used was accurate-mass, the precursor ion mass selection is unit mass or slightly greater, so two co-eluting compounds with the same nominal mass will both be fragmented and their spectra combined.
The Excel Add-In and the Rational Numbers search program were of little help in this particular case. This problem of mixed spectra is usually detected by looking at background spectra or by running blanks. Interfering ions can be subtracted out if there is sufficient chromatographic separation. This problem can often be quite subtle. For example, I often observed a 149 ion (fragment of the ammonium adduct of dioctyl phthalate, 390 daltons) in the MS/MS spectrum of a 408 MH+ compound that I worked with. While phthalates and their adducts often interfere in positive ion mode, watch out for stearate and palmitate ions, their acetate and formate adducts, and their dimers in negative ion mode.
The Excel Add-In sometimes catches convoluted spectra because it checks the data at the beginning of every analysis. The convoluted spectra will often have an unlikely mass loss. For example the 46.0193 loss in this example could be C2H3F, but often the loss may not match any formula because it is not derived from the assumed precursor.
"Extraterrestrial Ions"
Someone (name withheld) once wrote that to demonstrate that you have correctly identified a compound from its mass spectrum, you should be able to assign every fragment ion in its spectrum. Anyone who has actually worked on identifying unknowns from mass spectral data would realize that this is much too high a standard. First of all, life is short and unknowns can be identified with reasonable certainty without going to extraordinary lengths. Second, quite often spectra are not really pure and may contain extra ions from co-eluting compounds. Third, some spectra will have noise peaks that are impossible to explain; this is actually fairly common. The spectrum of interest here is the positive ion mode MS/MS spectrum of isopentenyladenosine found in massbank.jp that has four "extraterrestrial ions". When the Excel Add-In starts up, it checks the data in the blue columns to make sure, for example, that the data is numeric. But another check is for "extraterrestrial ions". These are ions in the spectra with mass defects such that no combination of the naturally occurring 92 elements (singly charged) can rationally explain the numbers. The Excel Add-In uses a function called ET() to check the defects of each entered mass before the analysis starts. If the mass is extraterrestrial, it will be then flagged and that data point should then be cleared. From the Excel Add-In results, the comment sheet lists the raw data where the red-colored ions have mass defects that are too high. The second worksheet illustrates the "cone of extraterrestrial mass defects". The remaining fragment ions were then successfully analyzed with the Excel Add-In and demonstrated excellent accuracy, indicating that the four extraterrestrial ions were indeed some kind of random noise. Note that extraterrestrial ions seem to occur with every type of instrument; perhaps a sign the instruments might need some maintenance. In this particular spectrum the remaining ions all appear to be both real and very accurate. The concern is that random ions, if their masses were found outside the cone of extraterrestrial mass defects, would be construed as real fragments arising from the unknown compound, thus making identification work more difficult.
The search software found isopentenyladenosine as the best match (results here) despite the presence of the four extraterrestrial ions. Run time was 2 to 3 seconds for pattern matching, 119 seconds for the Excel Add-In.
|

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}